| Issue |
BIO Web Conf.
Volume 17, 2020
International Scientific-Practical Conference “Agriculture and Food Security: Technology, Innovation, Markets, Human Resources” (FIES 2019)
|
|
|---|---|---|
| Article Number | 00247 | |
| Number of page(s) | 5 | |
| DOI | https://doi.org/10.1051/bioconf/20201700247 | |
| Published online | 28 February 2020 | |
Analyzing the exome heterogeneity of cattle immunity genes with the method of flow-cell sequencing
1
All-Russian Research Institute of Animal Breeding, Lesnye Polyany 141212, Moscow region, Russia
2
Ernst Russian Research Institute for Animal Husbandry, Dubrovitsy 142132, Moscow region, Russia
3
Institute of Animal Science, Pratelstvi 815 104 00 Praha Uhrmeves, Czech Republic
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Abstract
Toll-like receptors belong to the pattern-recognition receptors (PRRs), which have evolved to recognize conserved features of bacterial and viral molecules. We used the approach developed earlier to screen for the polymorphism in TLR genes in a representative set of historical and modern cattle breeds from Russia. The method pipeline included the steps of obtaining the overlapping amplification products from the coding regions of all ten bovine TLR genes, their subsequent purification and normalization. While the anti-bacterial group included TLR1, -2, -4, -5 and -6, the anti-viral group compressed TLR3, -7, -8, -9 and -10 (in spite of its unclear specificity). Animals from the about seven breeds, both bulls and cows, was used for analysis. The samples from the pooled genomic DNA were sequenced on the PacBio platform. After identification of variations, Bayesian analysis was carried out, followed by filtration on quality of sequencing. The 5–36 structural variants of TLRs were annotated according to their biological significance. Both new and already identified sites of variability, already annotated and documented in dbSNP, have been found. The data are needed for further breeding of local breeds in Russia with respect to their natural resistance to various diseases.
© The Authors, published by EDP Sciences, 2020
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License 4.0, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License 4.0, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
The polymorphism in bovine genes TLR1-10 in a set of local breeds of Russia was investigated. TLR genes code for Toll-like receptors, which represent a key part of ten system recognizing microorganisms and viral nucleic acids associated with bovine diseases and disorders. Among others, they are known to activate the signalling pathways that involve the interferon regulatory factors (IRFs), as a family of transcription factors known to play a critical role in antiviral defence, cell growth and immune regulation.
Three IRFs (IRF3, IRF5 and IRF7) functioned as direct transducers of virus-mediated TLR signalling, and their activation depends on the TLR stimulated: thus TLR3 activates IRF3 and IRF7, while TLR7 and TLR8 activate IRF5 and IRF7. In addition to these IRFs, several kinase-pathways are involved in fine-tuning the intracellular response.
Toll-like family (TLR) are essential in the formation of the innate immune response of animals to various pathogens, for example fungal, bacterial and viral origin [1]. The variability of immunity genes with high-throughput technology has been studied and shown to be a modern and real, and cattle immunity genes in part in genes TLR-1-10 that recognize viral nucleic acids, microorganisms and other biological molecules (polysaccharides, proteins) [2, 3].
It is also known that the variability of TLR genes can affect the predisposition of cows to complications during calving in the prenatal and post-partum periods. Such sequels can be caused by complications in the course of viral infections of respiratory paths or bacterial infections of the genitals and urinary tract.
In this case, a more severe course of infections can lead to fetal damage after calving, a more difficult pregnancy and premature abortions. The diversity of the Toll-like receptors are of direct relevance to the selection of highly productive animals demonstrating high disease resistance in life and disorders of the reproductive.
The obtained data are also expected to be used in the implementation of conservation programs for native breeds in the diverse regions of Russia.
2 Material and methods
The tissue samples were taken from cows and bulls of Kholmogorskaya breed (Pechora type, North type), Yakutskaya breed, Yaroslavskaya breed, Simmental dairy and meat / breed variant, Black-white breed, and the cattle – forest buffalo hybrid. Altogether, 575 samples of 14 populations were used for the study.
DNA from the sperm and cartilage specimen was hydrolysed with proteinase K and after 8-hour incubation, DNA was isolated by separation of the MagSep magnetic particles using the robotic station epMotion5070 (Eppendorf, Germany).
The DNA concentration was determined by UV-spectrophotometry and its integrity was checked electrophoretically.
The coding sequences of the TLR genes were amplified in a series of PCR reactions performed on an eqimolar DNA pool. Subsequently, an equimolar combined sample of all amplicons (31 of anti-bacterial TLRs, 52 of anti-viral TLRs) was prepared according to the electrophoretically determined yield of PCR reactions and the product molecular weight.
The pooled amplicon samples were purified using a NucleoSpin column (Macherey-Nagel, Germany). After additional purity checks with gel and Agilent chip electrophoresis, the amplicons were sequenced using SMRT sequencing technology (Pacific Biosciences, USA) on the RS-II instrument in the GATC Biotech (Constanz, Germany) sequencing center.
The average coverage for sequencing were up to 76 reads from 400 to 1200 bp in length, with a depth of coverage of 3–12 per individual.
Mutation testing was carried out using the SNAPshot methodology in a 3130xl capillary sequencer with primers that were specific for giving variations (Applied Biosystems, USA).
3 Results and discussion
TLRs are an evolutionary conservative family of mammalian molecules that recognize conservative patterns of microbial and viral structures. As such, they are absolutely necessary to determine strategies for protecting the host organism from pathogens.
In addition, TLRs play an important role in maintaining tissue haemostasis, for example, in healing of damaged tissues. TLR-1, TLR-2, TLR4, TLR-6 and TLR-10 are involved in lipid recognition, TLR-5 and TLR-11 recognize proteins, and TLR-3, TLR-7, TLR-8 and TLR-9 recognize nucleic acids [1, 3].
In mammals, TLRs are located on the membrane, in the endosomes, or in the cytoplasm of cells, and spends out the first line of defence (neutrophils, macrophages, dendritic cells, etc.). After ligand binding, TLRs trigger a signalling cascade involving a number of adapter proteins, which leads to the activation of nuclear factors and subsequent production of cytokines and other molecules associated with inflammation. The TLR signalling pathway is controlled by various feedback mechanisms.
In general, the biological role and mechanisms of TLR functioning are not fully understood. Today, the variability of immunity genes is being studied using highly efficient sequencing. The genetic diversity of TLR is directly related to the selection in the process of selection of highly productive animals, which also demonstrate high resistance to disease.
The previously developed approach was used for screening polymorphism in TLR genes in a sample of native and modern breeds of the Czech Republic for animal breeds from Russia [1]. The antibacterial group of genes included TLR 1, -2, -4, -5 and -6, and the antiviral group: TLR3, -7, -8, -9 and -10.
Identified structural variants of TLRs were annotated according to their biological significance. New and already identified variation sites were found, annotated and documented in the dbSNP database of the 1000 Genomes project and new synonymous variants (Table 1), while the number of synonymous substitutions in the genes was different from each other.
The primary processing of our data was carried out according to the flowchart implemented in the UGENE software package (v. 1.23.1, www.ugene.net) [4].
The quality assessment was performed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads of appropriate quality were aligned to the reference genome Bostau6 (https://genome.ucsc.edu) using BWA-MEM (www.bio-bwa.sourceforge.net/). After removing duplicates in Cigar MDWMC (http://broadinstitute.github.io/picard/), SNVs were detected using FreeBayes (https://github.com/ekg/freebayes) and SAMTOOLS (www.htslib.org/) (Tabl. 1–2, Fig. 1).
Figure 1 shows a diagram of a sequential algorithm for working with primary data to obtain the result in the form of SNV. The scheme provides for verification of the obtained data and it genotyping by capillary electrophoresis.
For further work, it is possible to use the AmpliSeq genomic libraries (https://goo.gl/iFJVeR, https://goo.gl/gRM4Xr) and TruSeq Custom Amplicon (coverage 92.8, 95%, amplicon length 150, 175 bp, total 331, 273 pieces, respectively) [https://goo.gl/zj6nTn, https://goo.gl/Q8ANb4) \ using the original chemistry or Kappa polymerase (Sigma Eldrich, USA).
The characteristics of the obtained sequence were analysed and the main data on the success of the experiment were calculated using the BAM file visualization module. This module is part of the UGENE software package (Table 1).
The primary characteristics of the sequencing performed showed a high level of genome coverage. Moreover, the amount of coverage reaching values of the order of 32,000 is due to the peculiarities of sequencing on the PacBio instrument. During sequencing, the reading process is repeated many times by repeatedly pulling the ring molecule through the flow-cell. In this case, the molecule also contains multiple sequences of amplicons, obtained using the same pairs of primers.
When determining such a characteristic as the number of reads, the factor value was tied to the coverage value and amounted to 550,000. This was also due to repeats in the sequencing process.
Further duplicates of amplification were removed, which reduced the weights of these parameters to reasonable values. Do not forget, that the amount of coverage depends on the initial concept of setting up the experiment with combining amplicons of different individuals in a single equimolar pool.
After cleaning the data on the quality of the readings and removing duplicates, the reads per individual reached 5.2–15.9 (Table 2). This amount of genome coverage for each individual was sufficient for an initial assessment of genetic heterogeneity.
The application of the FreeBayes algorithm in combination with SAMTOOLS allowed in an automatic mode to identify meaningful variants of mutations “in the flow”.
Only after the completion of these stages, the found single nucleotide variation (SNV) will be used for the association studies with the disease resistance/ susceptibility traits (Table 3). Application of the algorithms for factor statistics implemented in the SAS package (www.sas.com/) and the definition of haplotypes according to Patel et al. [5] will presumed.
It should be noted that the number of SNVs that were detected using the SAMTOOLS method was higher. This is due to the fact that this method, in comparison with the Bayesian method, has less strict settings for probabilistic search.
Summary and validation of the variant alleles obtained were carried out with the help of VeIP (www.eensembl.org). All identified SNVs were tested for compliance with biological significance, and also tested for errors and presence in the VeIP database (EensemblDB).
In this case, the probable variability of the amino acid sequence for the replacement of amino acids was checked if this could change the structure of the receptor molecule? Identified SNVs so should have been contained in the dbSNP database. Table 3 lists only those SNVs, that have passed all the verification steps described above.
It is important that the variations for the TLR1 and TLR6 genes was indicated together, since the exomic gene sequences overlaps. For the TLR4, 5, and 9 genes, many new variations have been identified. Perhaps such a high value of variability requires a separate study to clarify the accuracy of calculation algorithms and additional sequencing of this area.
This is necessary in order to exclude possible sequence errors that may arise due to the characteristics and quality of the process. PacBio technology is currently being improved and there may be errors or artefacts that may be due to failures of this new sequencing method.
In the next step of the work, the found genotypes will require validation in order to exclude errors that occur during poor-quality sequencing. Currently, the genotyping assays exploiting the primer extension technology as enabled by the SNaPshot Multiplex Kit (Applied Biosystems, USA) are applied to this task. The validated novel SNVs will be added to the NCBI dbSNP database.
Identified variations are presented. In the study, animals were genotyped individually according to 15 probable SNVs located on 4 different genes (Figure 2). Identified variations are indicated with a frequency characteristic throughout the mixed sample. Options for the study were selected those that were most likely to occur in the represented population.
Since genotyping was carried out individually, it generally reflects the frequency of occurrence of SNV in the representative group of cattle animals. Importantly, a number of SNVs presented did not confirm their variability, as suggested by sequencing data.
This is a predictable result, since it was planned that the detected variability could not be confirmed in the future by direct verification of mutations. This screening was the last stage after careful filtering of the detected SNVs at the previous stages of operation.
It should be noted that the analysis was not carried out for specific breeds separately. In the future, it is planned to expand the testing panel and a more detailed analysis of animals will be undertaken if this work finds funding in the future.
Parameters of the TLR genes resequencing
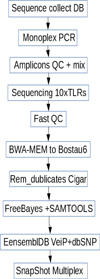 |
Fig. 1. Scheme of amplicons obtaining, quality control of data, data processing and SnapShot genotyping in capillary electrophoresis. |
Final characteristics of sequencing in the TLR genes
Variants indentified in the TLR genes
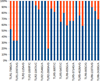 |
Fig. 2. Results of preliminary testing of gene variability using SNaPshot. SNP positions are presented according to FJ147090, EU746465, EU006635 and AJ618974 for TLR receptor genes 1, -2, -5 and -6, respectively. |
4 Conclusion
In conclusion, sequences of ten TLR genes of cattle from seven different breeds were obtained.
In conclusion, we note that the sequences of the exomic regions of ten TLR genes of cattle from seven different breeds of Russia were obtained. The method of ultra-long high efficient sequencing made it possible to identify up to 64 putative mutations in the coding sequences of the studied genes and also confirm 15 variations using the primer extension method. This information allows us to predict their further appearance in the population using methods of direct genotyping of bulls, for example using gene chips and real-time PCR.
This information allows for the determination of their occurrence in the population using direct genotyping assays.
The SAM TOOLS-method as a whole revealed more mutations in genes than the Bayesian method. Identified mutations require further assignment at the individual level to identify associations with the susceptibility to diseases.
We plan to refine animal diagnostics by the monitoring of the expression of immunity genes, as elicited in experimental groups of animals with external factors. The available techniques of parallel sequencing or real-time PCR will be applied according to the number of targets and the expediency of scaling, as exemplified in infected animals with different viral load [6–7].
Acknowledgements
The work was supported by the Program in the Research Institution Development of the Ministry of Agriculture of the CR no MZERO0714 and by National Program of Conservation and Use of Animal Genomic Resources.
References
- K. Novak, Vet. Immunol. Immunopathol. J., 157, 1–11 (2014) [CrossRef] [Google Scholar]
- R.G. Schaut, J.F. Ridpath, R.E. Sacco, PLoS One, 11, e0159491 (2016) [CrossRef] [PubMed] [Google Scholar]
- F.C. Mansilla, M.E. Quintana, N.P. Cardoso, A.V. Capozzo, Parasite Immunol. J., 38, 663–669 (2016) [CrossRef] [Google Scholar]
- K. Okonechnikov, O. Golosova, M. Fursov, Bioinformat., 28, 1166–1167 (2012) [CrossRef] [PubMed] [Google Scholar]
- S.M. Patel, P.G. Koringa, N.M. Nathani, N.V. Patel, T.M. Shah, C.G. Joshi, Meta Gene., 3, 50–8 (2015) [CrossRef] [PubMed] [Google Scholar]
- M.V. Farias, P.A. Lendez, M. Marin, S. Quintana, L. Martmez-Cueta, M.C. Ceriani, G.L. Dolcini, Res. Vet. Sci., 107, 190–195 (2016) [Google Scholar]
- D. Werling, J. Piercy, T.J. Coffey, Vet. Immunol. Immunopathol., 112, 2–11 (2006) [CrossRef] [PubMed] [Google Scholar]
All Tables
All Figures
|
Fig. 1. Scheme of amplicons obtaining, quality control of data, data processing and SnapShot genotyping in capillary electrophoresis. |
| In the text | |
|
Fig. 2. Results of preliminary testing of gene variability using SNaPshot. SNP positions are presented according to FJ147090, EU746465, EU006635 and AJ618974 for TLR receptor genes 1, -2, -5 and -6, respectively. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.